- 0
-
找原料、比价格、代采购找我!
 前小衍平台客服最近30天成交374单027-81293128
前小衍平台客服最近30天成交374单027-81293128
 产品合作与推广,清库存找我!
产品合作与推广,清库存找我! 唐令平台采购15072440602
唐令平台采购15072440602

-
您的随身业务助手
微信扫一扫 关注「前衍化学」公众号


 发询盘 看报价收询盘 拿订单
发询盘 看报价收询盘 拿订单
微信扫一扫 关注「前衍化学」公众号
发询盘 看报价收询盘 拿订单
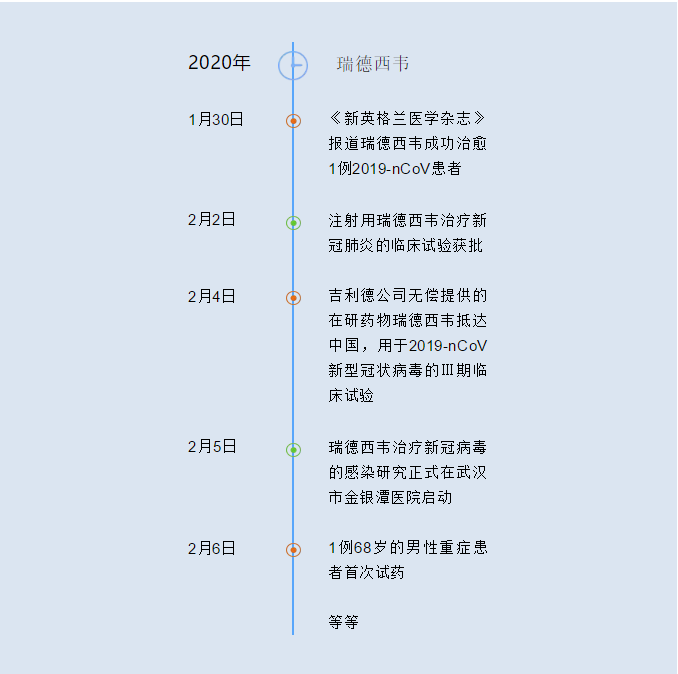
瑞德西韦成为在我国为数不多的跳过Ⅰ/Ⅱ期临床试验,直接进入Ⅲ期临床试验的药物,也是目前唯一一个Ⅲ期临床试验的药物。
武汉病毒学研究所探究了瑞德西韦对2019-nCoV的体外抑制活性,结果表明,作用机理与其他核苷类抗病毒药物一致,主要作用于病毒进入细胞后期阶段;瑞德西韦对感染2019-nCoV的Vero E6细胞抑制活性EC50=0.77 μmol·L-1、细胞毒作用CC50>100 μmol·L-1、选择性指数SI>129.87。对感染细胞的EC90=1.76 μmol·L-1。
这款被视为人民的希望的药物到底是怎么研发出来的呢?
1、先导化合物的发现
吉利德凭借其在抗病毒类药物研发领域积累的丰富经验和雄厚的技术实力,2010年开始着手研究抗埃博拉病毒的药物。
研究人员在P4实验室筛选了1000多个核苷类化合物对埃博拉病毒感染的人微血管内皮细胞(EBOV- HMVEC)的抑制活性,发现衍生物1抑制活性(EC50=0.78 μmol·L-1)最好。
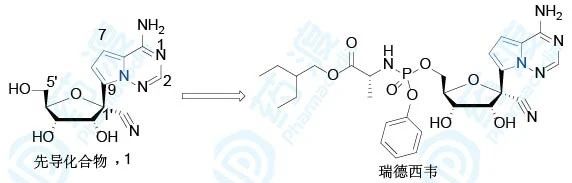
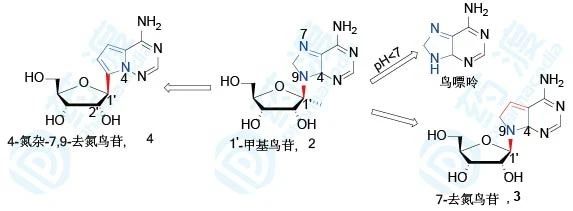
核苷类药物很少在戊糖的1′位进行取代,主要原因是其化学稳定性比较差。如1′-甲基鸟苷(2)在pH<7的溶液中N-C苷键断裂,释放出鸟嘌呤,但是研究发现,戊糖与核苷以C-C键相连时则稳定较好不易断裂。另有研究显示,7位氮原子去掉后,得到结核菌素(3,7-去氮鸟苷),结核菌素三磷酸化后是一种RNA依赖的RNA聚合酶底物,可参入到RNA链中,表现出较强的细胞毒性。但是4-氮杂-7,9-去氮鸟苷(4)细胞毒性消失而且具有与衍生物3相当的抗肿瘤活性。所以,研究者猜想,以衍生物1为先导化合物,这种C-C键连接戊糖和核苷的模式作为抗代谢物具有一定的可行性。
2、结构修饰
2.1 碱基环修饰
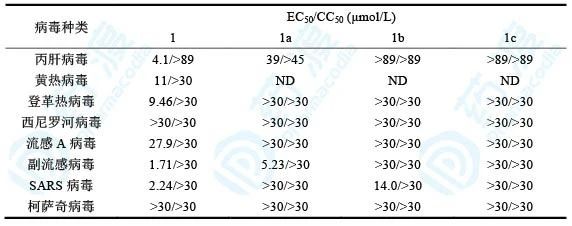
研究者利用甲基、乙烯基和乙炔基取代化合物1的1′-氰基,并测定了这些化合物对丙肝病毒、黄热病毒、登革热病毒、西尼罗河病毒、流感A病毒、副流感病毒、SARS病毒和柯萨奇病毒的抑制活性和安全性。
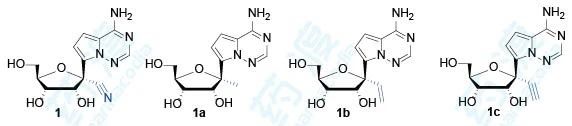
表1 4-氮杂-7,9-去氮鸟苷类衍生物对常见病毒的抑制活性
研究者进一步探究了化合物1的1′和2′取代基的变化,以及碱基环的变化对已感染埃博拉病毒(EBOV)、呼吸道多核体病毒(RSV)和丙肝病毒(HCV)细胞的半数抑制浓度EC50,同时也评价了这些化合物对正常细胞的半数抑制浓度CC50,探究其安全性。
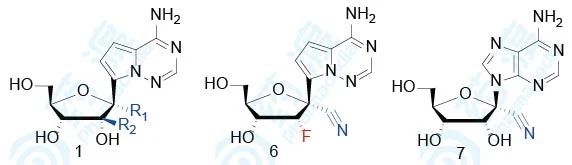
表2 核苷类衍生物生物活性和毒性(a是HMVEC细胞、b是HEp-2细胞、c是Huh-7细胞)
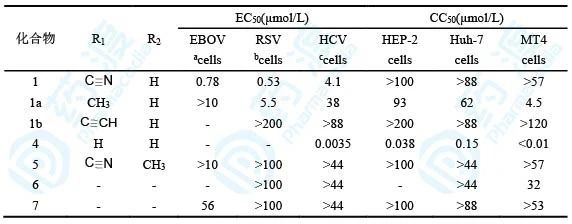
从这里可以看出,1′-氰基化合物抗病毒活性好且细胞毒性小;1′-甲基化合物和1′-乙炔基化合物抗病毒活性消失;1′-氰基-2′-F化合物以及碱基换成鸟嘌呤抗病毒活性均消失。这些结果表明,化合物1在抗病毒活性和选择性上都呈现最优状态。
故研究者直接将化合物进行三磷酸化得到衍生物8,进一步测定其对病毒和人体RNA依赖的RNA聚合酶(RdRp)的生物活性。
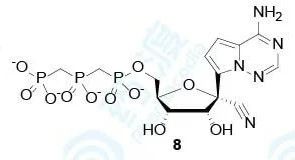
表3 化合物8对RSV聚合酶、HCV聚合酶和人类聚合酶的抑制作用

注:SNI rate=single nucleotideincorporation rate(单核苷酸结合速率)
从这里也可以看出,化合物8对RSV聚合酶和HCV聚合酶具有较好的抑制活性,与化合物1的生物活性吻合。由于此时,并没有从埃博拉病毒中提取出聚合酶,所以通过测定细胞连续动态估算出化合物8对埃博拉病毒聚合酶的抑制活性与丙肝病毒聚合酶抑制活性相当(IC50≈5 μmol/L)。另外,化合物8对人体线粒体聚合酶和RNA/DNA多种聚合酶的抑制活性均较差(IC50>200 μmol/L),且向RNA单链中的参入很低,说明化合物8对病毒选择性高。
2.2 侧链修饰
由核苷类药物的抗病毒机理可知,核苷类药物参入到RNA链中抑制病毒复制,需要经过三磷酸化过程。其中第一次磷酸化是限速步骤,这一步对病毒的抑制作用强度影响最大,所以研究者将第一次磷酸化设定在戊糖的5′-位,既往研究均表明了这种方式的磷酸化有利于第二次磷酸化和第三次磷酸化的发生,从而发挥出抗病毒的活性。但是进行一次磷酸化后的产物含有两个负电荷,化合物的亲水性增强,不利于衍生物的跨膜转运和细胞内的传输。在研发抗丙肝药物索非布韦(9)时,研究者也遇到了类似的困境,但是将其制备成磷酰氨酸酯前药后,取得了重大突破,故此时研究者也参照了这种处理方式,制备了相应的磷酰氨酸酯前药,用于掩蔽负电荷的极性,设计合成了一系列化合物,并测定了衍生物对易感染埃博拉病毒(EBOV)、呼吸道多核体病毒(RSV)和丙肝病毒(HCV)细胞的半数抑制浓度EC50,同时也评价了这些化合物对正常细胞的半数抑制浓度CC50,作为初步安全性指标。
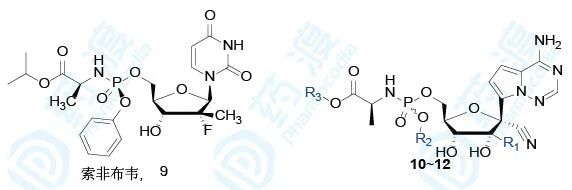
表4 核苷类衍生物生物活性和毒性(a是HMVEC细胞、b是HEp-2细胞、c是Huh-7细胞)
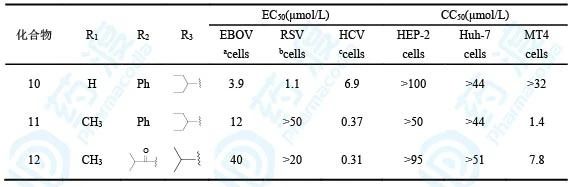
从这里可以看出,化合物10对埃博拉和呼吸道综合征病毒的抑制活性强11和12,这也与前面的核苷类衍生物1和5之间的生物活性差异相符合。衍生物10对人体正常细胞的作用很弱,提示有较高的选择性。值得提及的是11和12对丙肝病毒的活性显著强于10,这是因为2′-甲基取代更有利于与丙肝病毒聚合酶的结合,索菲布韦的戊糖片段就是2′-CH3-2′-F取代。分子模拟也显示2′-CH3取代有利于同HCV的结合,而不利于同EBOV和RSV聚合酶的结合。
2.3 磷酰氨酸酯修饰
随后研究者对磷酰氨酸酯结构进行修饰。设计合成了一系列化合物,并测定了其生物活性。
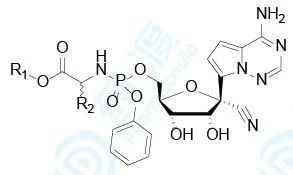
表5 磷酰氨酸酯前药的抗病毒生物活性、半衰期和logD
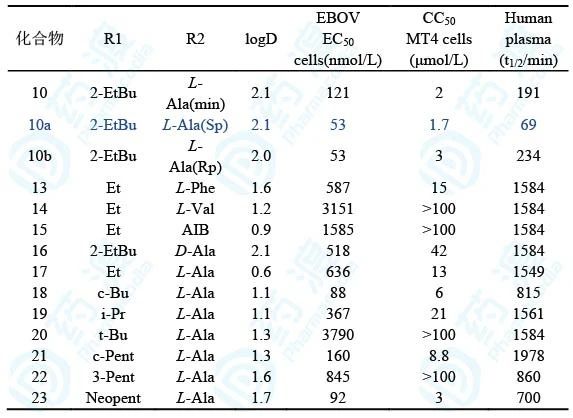
从这里可以看出,(1)由于磷酰氨酸酯的磷原子为手性中心,前药10是 1:1的差向异构体混合物,经分离得到纯净的S和R异构体10a和10b,抑制埃博拉病毒的活性EC50均为53 nmol/L,都高于混合物10,原因不详。后来选定10a作为候选化合物的原因是具有广谱抗病毒活性和制备过程结晶性较好的缘故;(2)L-Ala换成D-Ala活性显著降低,提示L构型的必要性。L-Ala换成其他氨基酸,例如L-Phe、L-Val和2-氨基异丁酸(AIB)的活性都降低,因而氨基酸片段优选为L-Ala;(3)末端酯基的大小和形状对活性有影响,乙基(17)、异丙基(19)、叔丁基(20)和3-戊基(22)活性降低;而环丁基(18)、环戊基(21)和新戊基(23)的活性与2-乙基丁基(10)相近。
3、候选药物确定
10a的药代动力学研究是在猕猴身上进行的,一方面是由于猕猴的病理生理学与实际的人类疾病相似,另一方面是啮齿类动物血浆中含有高表达的酸酯酶,导致10a快速进行血浆代谢。
用猕猴进行药代动力学实验表明,口服给药后首过效应明显,生物利用度低,而且感染埃博拉病毒后的患者不宜口服用药,因此最终选择静脉滴注的方式给药。
猕猴静注10a后,10a在血液中迅速分解为一磷酸核苷,并且在2h内迅速分布在外周血单核细胞中,最终形成三磷酸核苷(8)发挥药效。比如,猕猴静注10 mg·kg-1的10a后,血浆中8的浓度是体外抑制埃博拉病毒IC50的数倍,且可维持24 h,半衰期长达14 h,故推测每日给药一次即可维持有效治疗水平。
在I期临床试验中,对志愿者进行静脉注射10a后表现出良好的药代动力学性质,且副作用少,将剂量由150 mg增大到225 mg也未见明显的不良反应,对成人和婴儿患者的治疗14天后血液和脑脊髓中埃博拉病毒消失。故将10a命名为瑞德西韦(remdecivir)。
在II期临床试验时,此时与瑞德西韦同台竞争的药物有三个。在第一批患者测试中,瑞德西韦的表现并不佳,53%患者接受治疗后死亡,mAb-114、REGN-EB3和Zmapp的死亡率分别为34%、29%和49%。若患者血液中的埃博拉病毒水平不高,使用REGN-EB3后,存活率可高达94%,mAb114也有89%,ZMapp为76%,但是Remdesivir的存活率仅为67%,从而终止了临床试验。
至于瑞德西韦是否真能成为人民的希望、2019-nCoVs的克星,还须耐心等待正在进行两项Ⅲ期临床试验结果,但是瑞德西韦在治疗埃博拉病毒上的临床试验至少证实,安全性较好。
我们期待临床试验能够取得一个良好的疗效!!!
参考资料
[1] Remdesivir and chloroquine effectively inhibit therecently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res, 2020, 0:1-3.
[2] Discovery and synthesis of a phosphoramidateprodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) forthe treatment of Ebola and emerging viruses. J Med Chem, 2017, 60: 1648−1661.
[3] 治疗沙粒病毒科和冠状病毒科病毒感染的方法.
关键词:COVID-19 瑞德西韦 研发历史
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2024 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1

